近日,我院汤富杰副教授与嘉庚创新实验室AI4EC Lab团队利用机器学习联用方法,在计算电解液动态核磁共振(NMR)谱解析领域取得重要突破,实现了对双氟磺酰亚胺锂(LiFSI)/二甲醚(DME)电解液中动态的⁷Li核磁共振化学位移的预测。预测结果精准揭示了⁷Li核磁共振化学位移的反转现象,与实验观测结果吻合。该成果是团队继电池正极材料动态核磁谱研究和NMRNet深度学习框架之后,在相关领域的又一次方法迭代与体系应用拓展,相关研究成果已发表于顶级化学期刊《Journal of the American Chemical Society》。该项研究成果的通讯作者为化院程俊教授和我院汤富杰副教授,厦门大学化学化工学院2023级博士研究生尤祺和博士后孙岩为共同第一作者,嘉庚创新实验室副研究员王锋在研究过程中提供了协助。
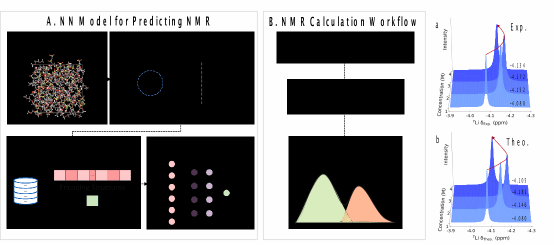
(左)训练神经网络(NN)模型的方法。捕捉不同浓度下的各种结构,并提取Li⁺的溶剂化结构。随后,使用描述符对结构进行编码,并计算其相应的化学位移。(中)NMR谱预测流程。使用机器学习分子动力学(MLMD)模拟生成轨迹,然后利用所得到的核磁共振预测神经网络模型来获取核磁共振光谱。(右)NMR谱预测结果与实验结果的对比。
核磁共振(NMR)谱无损且对局域结构敏感,能表征溶剂化结构中特定原子核化学环境,揭示弛豫时间等信息,但将谱变化与分子结构变化联系极具挑战。一些采用密度泛函理论(DFT)计算的研究,通过静态计算NMR谱来解释结构-光谱关系。然而,实验化学位移反映的是来自不同局域位点的加权平均,融合了局域结构和动力学信息,这种统计平均使得信号分辨变得复杂,也加大了解析谱构关系的难度。分子动力学(MD)模拟可捕捉电解质动态结构变化,但在复杂体系中构型采样复杂,难以将分子结构与实验光谱观测值直接联系,虽有机器学习模型提高化学位移预测速度,但目前电解质动态结构特征与实验光谱观测值的关系尚无明确共识,亟需有效的计算方法,且验证模拟与实验关联对可靠性至关重要。
基于此,团队提出了一种基于机器学习联用的方法,来预测LiFSI/DME电解质溶液中动态的⁷Li核磁共振化学位移,预测结果揭示了⁷Li核磁共振化学位移的反转现象,与实验结果吻合。LiFSI浓度从1 M增至3 M时,溶剂化结构变化使NMR化学位移向高场移动,4 M时则向低场移动。此外,团队定量地建立了分子结构与核磁共振谱之间的关系,深入剖析了溶剂化结构的归属。分析表明,存在两种相互竞争的局部溶剂化结构,电解液浓度接近上限时,其主导地位交替,导致⁷Li化学位移变化。该方法精准高效,有望拓展到其他复杂电解质体系,并预测其他原子核化学位移。这项工作为深入理解核磁共振谱所探测到的复杂溶剂化结构提供了分子层面的详细认识,为改进电解质的设计指引了方向。
该研究工作受到国家自然科学基金(资助号:22021001、22225302、21991151、21991150、92161113)、中央高校基础研究基金(资助号:20720220009)、国家重点研发计划(资助号:2024YFA1210804)以及人工智能应用电化学实验室(AI4EC)、IKKEM(资助号:RD2023100101和 RD2022070501)的资金支持。
论文链接:
●https://pubs.acs.org/doi/10.1021/jacs.5c02710
